




中国创新药国际化进阶的机遇与挑战——信达/礼来OADC会议涉及的国际临床研究问题分析
自2021年1月1日至2021年12月31日期间,共有3247项药物临床试验在中国药物临床试验登记和信息公示平台进行了公示,同比增加了2.7%,排名前20的适应症多为肿瘤和慢性疾病及精神类疾病,同时肿瘤专科医院以及肿瘤方面较强的综合医院进行临床试验的数量也位居前列,体现出我国抗肿瘤创新药物研发的蓬勃发展。快速发展的同时也带来了竞争和拥挤,行业都在思考下一步应该往哪里走。从2021年7月2日国家药品审评中心在其官网发布的《以临床价值为导向的抗肿瘤药物临床研发指导原则(征求意见稿)》一稿引发千层浪,到北京时间2022年2月10日信达/礼来PD-1单抗的肿瘤药物咨询委员会(ODAC)会议直播4万多人在线观看被戏称为“中国医药界的春晚”(笔者亦旁听了该直播),行业内对于中国创新药发展之路的探讨从未停止。结合近些年来国际社会在药物研发领域的新共识以及新探索,我们希望通过本文对创新药出海国际化道路上单一海外研究数据在美国的适用性问题进行梳理和分析,以期阐述中国创新药国际化进阶之路上的机遇与挑战,对未来药物研发以及上市的监管方向作出预判。
本文共计5000字,建议阅读时间为10分钟。
北京时间2022年2月10日晚11点,ODAC以在线方式召开会议,会议就信迪利单抗(sintilimab)针对非鳞状非小细胞肺癌的新药上市申请进行了公开讨论(“ODAC会议”),与其说议程的焦点是中国临床研究数据对于美国人群的适用性问题,不如说ODAC会议希望通过这种方式在充分的公众视野下阐述并试图建立新的规则。当然,需要强调的是ODAC一般以会议讨论的方式,就药品的安全性、有效性等存在争议的问题,为美国食品药品监督管理局(FDA)提供专业性意见,其意见会影响新药的审批过程,FDA做决策时会采纳相应的意见,但ODAC对FDA并没有法律约束力,FDA并非一定要采用ODAC的建议,从历史记录来看,ODAC和FDA意见存在不一致的情况。所以在目前阶段只能说ODAC阐述了其对于《美国联邦法规》(Code of Federal Regulations, CFR)采纳单一海外研究数据以及其适用性问题的进一步理解。以下就会议中涉及的几个关于国际临床研究问题分别展开阐述。
一、美国CFR对于海外临床试验数据的接受要求
根据21CFR312,在满足一定前提的条件下,FDA接受一项设计良好、进行良好的海外临床研究,作为申请上市批准的支持,这些前提条件包括:(1)临床试验依照良好的临床试验质量管理规范(good clinical practice, GCP)进行(包括符合对于遵守伦理的要求)[1];(2)如果FDA认为有必要,可以通过现场检查来验证研究数据[2]。进一步地,21CFR314规定了仅基于符合美国上市许可标准的海外临床数据的申请批准条件:(A.)海外数据适用于美国人口以及美国医学实践[3];(B)研究必须由具有公认能力的临床研究人员进行[4];(C)海外数据在无需FDA进行现场检查的情况下被认为是有效的,如果FDA认为有必要检查,FDA可以通过现场检查或其他适当方式来验证研究数据。[5]FDA同时为该等法规的适用提供了较大的弹性空间,明确了FDA将根据药物的性质和所考虑的数据以灵活的方式适用该政策[6],也就是说在仅提供海外临床数据在美国进行上市批准申请的情况下,FDA具有较大的自由裁量权。同时,FDA也鼓励申请人在申请前征询FDA的意见。
从会前资料以及会议上的讨论可以看出,双方花了较多时间针对本申请中的海外数据是否适用于美国人口以及美国医学实践进行了讨论,重点包括,(1)ODAC认为无法代表美国的种族多样性,ODAC会议提及“接受不反映美国人口多样性的单一国家外国数据挑战了行业对患者公平和纳入代表性不足人群的广泛承诺”;以及(2)试验设计与美国现有的医疗标准之间的差距,应以总生存期(Overall Survival,OS)而非实际所采用的无进展生存期(Progression-Free Survival,PFS)作为终点。虽然双方就研究人员的资质以及被自媒体广泛提及的2016年中国药监局报告中的数据不合格问题进行了一些讨论,但是就本申请中的临床试验数据的有效性以及真实性没有提出异议。
二、ICH E5以及ICH E17
与适用性问题相关的另一个延伸性话题是ODAC提及认为需要根据ICH E17的要求进行国际多地区/多中心临床试验(MRCT)而ICH E5不是一个理想的方法,而这可能导致申请人需要花费较多的费用以及较长的时间。
国家药监局于2019年11月5日发布关于适用《E1:人群暴露程度:评估非危及生命性疾病长期治疗药物的临床安全性》等15个国际人用药品注册技术协调会指导原则的公告,公告自该日起适用《E5(R1):接受国外临床试验数据的种族因素》及《E5问答(R1)》和《E17:多区域临床试验计划与设计的一般原则》等15个国际人用药品注册技术协调会(ICH)指导原则。
《E5(R1):接受国外临床试验数据的种族因素》(E5(R1)Ethnic Factors in the Acceptability of Foreign Clinical Data)制定于1998年,提供了协调和发展的策略,在尽可能减少重复临床研究和为使病人受益迅速提供药物的同时充分考虑种族因素的影响:国外临床资料可能符合新地区的要求时,推荐采用国外临床资料。但E5(R1)同时提及一旦临床资料符合新地区的管理要求,这些资料是否可以被采纳取决于新地区的人群沿用,如果管理当局关注在新地区人群种族因素可能改变药物的安全有效性时,申请人必须在新地区做出一定数量的临床资料以使得临床资料在两地之间可以延用和跨越,比如被设计成桥接试验(bridging study)。从中国的监管路径来看,根据《国家药品监督管理局关于发布接受药品境外临床试验数据的技术指导原则的通告》,境外临床试验数据应支持有效性和安全性评价,药品注册申请人应考虑符合中国药品注册管理要求,在对完整临床试验数据包分析的基础上,对关键临床试验数据进行评价,以确证研究药物的有效性;遵循ICH关于接受国外临床资料的种族影响因素(E5)要求,分析中国亚组与总体人群的一致性,以支持境外临床试验结果外推至中国人群。
相对于E5(R1)的较早形成,ICH于2014年6月启动了多地区临床试验(E17)指导原则的起草工作,并于2016年6月形成初稿。经过讨论后,最终于2017年11月通过《E17:多区域临床试验计划与设计的一般原则》(E17 General Principle on Planning and Designing Multi-Regional Clinical Trials),该指导原则主要阐述了MRCT的整体规划和设计的总体原则,以提高MRCT在全球监管机构中的认可度,增强MRCT在全球注册申报过程中的可接受性。同步地,2015年1月30日,国家药品监督管理局发布了《关于发布国际多中心药物临床试验指南(试行)的通告》,明确申办者要根据早期研究数据、种族敏感性分析和不同监管机构的要求,确定在全球不同区域间应采用的临床试验方式。
对于ICH E17以及ICH E5的规则适用,ODAC在会议中提出其认为桥接试验无法解决有关的普遍性问题,从1990年代后期到2017年之间ICH的演变已经表明了国际监管机构不再认为桥接试验是一种理想的方法,为了提供药物开发效率同步在全球获得批准,建议采取国际多地区/多中心临床试验,且针对目前递交的各申请,FDA采用ICH E17来进行审评。同时,考虑到FDA的检查和数据验证范围有限的问题,FDA会更加倾向于强调国际多地区/多中心临床试验。虽然在会议中申请人较为明确的阐述了其只是为了在中国批准而进行了临床,后来认识到使用中国数据可以在美国申报这个途径,所以进行了申报,但是从会议中可以看出ODAC依然倾向于采用国际多地区/多中心临床试验的数据,如果希望可以在全球或在多个国家或地区上市,可能需要尽早与各地的监管当局沟通,并进行国际多地区/多中心临床试验布局。
三、FDA药品上市许可批准先例
从FDA审批的先例来看,2017年FDA批准了治疗渐冻症(Amyotrophic Lateral Sclerosis,ALS)的药物RADICAVA,从FDA公布的信息[7]来看,FDA根据对368名ALS患者进行的三项临床试验的证据批准了RADICAVA,这些试验均在日本进行,无论是安全性还是有效性的临床试验。FDA亦明确了所有患者均为亚裔(图表一);因此,无法确定不同种族对RADICAVA的反应差异[8]。据悉,美国在该领域多年来没有新药上市,治疗手段停滞不前,因为FDA主动邀请企业进行上市申请[9]并完成了加速批准。
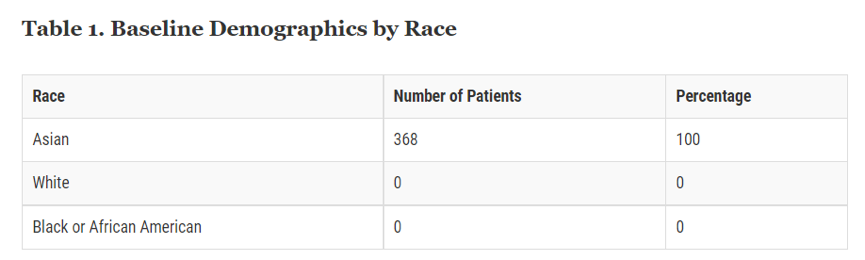
图表一
其他可以参考借鉴的当属百济神州的BTK抑制剂泽布替尼获FDA加速批准,泽布替尼的最终获批是基于两项临床试验的有效性数据[10],一为在中国开展的单臂Ⅱ期临床研究BGB-3111-206,一为在澳大利亚开展的全球Ⅰ/Ⅱ期临床试验BGB-3111-AU-003。虽然泽布替尼获批主要基于中国的研究数据,但在澳大利亚进行的BGB-3111-AU-003研究不只证明了泽布替尼在更多人群中疗效不错,还起到了桥接的作用:因为澳大利亚入组了很多高加索人种,在这个人群中同样看到了很好的疗效和安全性。
四、监管口径以及全球发展趋势
如上文第一部分所述,基于FDA具有较大的自由裁量权,除了正式的与FDA之间的征询外,创新药研发公司试图从ODAC/FDA官员公开发表的讲话以及文章中寻求ODAC/FDA的监管口径和态度,同时,可以预见到的创新药研发以及销售的全球发展趋势也从一定方面给创新药进阶同时带来机遇和挑战。
(一)FDA的弹性监管
FDA肿瘤卓越中心主任Pazdur从2019年AACR年会到ODAC开会前的2022年2月期间,态度的确发生了较大的转变。2022年2月4日(ODAC会议前6天),其在发表的“Importing oncology trials from China, a bridge over troubled waters”文章中讨论依据单一国家临床试验数据问题时提及对于确定该等数据的可接受性及其对于新人群的普适性方面,监管的灵活程度应当与药物的创新相平衡[11],比如是否有已经获批的产品、或者该等疾病是否在亚洲更常见,特别举例肝细胞癌或鼻咽癌,似在暗示对于该等药品若即使仅有单一国家或地区的临床试验数据,为了满足美国的医疗需求,也会有更高的接受度;或者可以解读为,如果是美国尚无上市药物的适应证或罕见病领域,仅有中国数据可能会有较高的获批几率。
(二)对于Me Better 和First in class药物的青睐
药物上市的根本目的是解决患者的需求,药物研发应以患者需求为核心,以临床价值为导向已经逐渐成为普遍共识。2017年FDA发布了以患者为核心的药物研发指导原则的制定计划,计划发布四项指南。2021年7月,药品监督管理局关于公开征求《以临床价值为导向的抗肿瘤药物临床研发指导原则》意见的通知对创新药企新药研发提出了更高的要求,不仅做Me-Too,而是需要致力于Me Better 和First in class的药物研发,一经问世,便在业内外引起广泛讨论。
2021年12月,《新英格兰医学》杂志上的一篇题为“The Wild West of Checkpoint Inhibitor Development”的文章中,FDA评论整个行业十分混乱,全球范围内在免疫检查点抑制剂(Checkpoint Inhibitor)类药物的快速扩张缺乏协调,造成了极大的医疗资源浪费[12]。从这个层面来看,会议中针对非劣效性研究的讨论中也听到了投赞成票的委员对于“不确定批准这种药物的紧迫性的理由” 的论点,意在暗示市场上的相同或类似产品较多,没有批准该等产品的紧迫性,所以也不用考虑临床试验需要做多久。基于此,也有行业专家预测,出于临床资源和竞争格局的考虑,FDA会对PD-1单抗的审批收紧。如果继续开发临床优势有限的Me Too类药物,需要大规模的资金投入开展符合各国监管要求的MRCT。更进一步说,可能预示着未来Me Too药物的研发投入和要求会更高,也需要更紧的跟随,否则将面临试验方案无法受到FDA 认可的情况。而对于未被满足临床需求的要求,FDA可能会给予更高的监管灵活性,也就是说Me Better和First in class的药物研发将更受青睐。
(三)价格因素
我国深化医改以来,国家采取一系列措施降低药品价格,新增17种抗癌药进入医保目录,药品集中采购和使用试点地区中标药品价格平均降幅达52%[13],政策红利正在惠及更多百姓,价格因素是医改的一个重要组成部分。虽然价格因素不是ODAC会议讨论的焦点,甚至不是一个议题,但是此次ODAC会议中委员对于价格问题的论述值得细细品味。根据外媒的报道[14],礼来承诺降低药价并可达到节省美国医疗系统数十亿美元的效果。投反对票的委员表示其不认为已经有了过多的抗癌药物,如果真的过多,就会看到价格下跌,而实际上并没有。ODAC虽然明确了FDA在做出监管决策时不考虑花费或药品价格[15],但FDA最终审批时是否会从一定层面上考虑到药品价格,或从药品价格降低可能带来的市场反馈出发影响到最终审批结果,我们需要拭目以待。
结语
此次ODAC会议对未来中国创新药出海具有一定的影响,这点对行业各参与方都具有警醒的意义。但作为一定特殊历史背景下的尝试事件,我们不认为此次事件一定是针对于大范围以及适用于未来所有中国创新药的结果,也不一定是FDA对于中国创新药企关门的态度信号。具体的美国监管动向有待进一步观察。在有限的医疗资源下,除了更好的协调统筹药品研发方向,各国都在考虑推动临床数据互认,促进发展多国平行审查计划(例如Project Orbis[16]),以期以更效率的方式推动药品上市,真正达到以临床价格和以患者需求为导向的药物研发。
【实习生何馨悦对本文亦有贡献】
注释:
[1] The study was conducted in accordance with good clinical practice (GCP).
[2] FDA is able to validate the data from the study through an onsite inspection if the agency deems it necessary.
[3] The foreign data are applicable to the U.S. population and U.S. medical practice.
[4] The studies have been performed by clinical investigators of recognized competence.
[5] The data may be considered valid without the need for an on-site inspection by FDA or, if FDA considers such an inspection to be necessary, FDA is able to validate the data through an on-site inspection or other appropriate means.
[6] FDA will apply this policy in a flexible manner according to the nature of the drug and the data being considered.
[7] Drug Trials Snapshots: RADICAVA | FDA,最后访问时间:2022年2月20日。
[8] All patients were Asian; therefore differences in response to RADICAVA among races could not be determined.
[9] “After learning about the use of edaravone to treat ALS in Japan, we rapidly engaged with the drug developer about filing a marketing application in the United States,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, FDA approves drug to treat ALS | FDA,最后访问时间:2022年2月20日。
[10] FDA grants accelerated approval to zanubrutinib for mantle cell lymphoma | FDA,最后访问时间:2022年2月20日。
[11] The degree of regulatory flexibility in establishing the acceptability of data from a single country and its generalisability to a new population should be balanced against the drug’s innovation. 摘自 Importing oncology trials from China, a bridge over troubled waters。
[12] Checkpoint Inhibitor Proliferation Like ‘Wild West,’ says FDA (biznewspost.com),最后访问时间:2022年2月20日。
[13] 我国推进药品领域改革 促进药品价格降低_国务院动态_中国政府网 (www.gov.cn),最后访问时间:2022年2月20日。
[14] Eli Lilly promises 40% discount for Innovent's PD-1 in last-ditch bid to shift FDA review to drug pricing | FiercePharma,最后访问时间:2022年2月20日。
[15] FDA does not consider cost or drug pricing in regulatory decision making.
[16] Project Orbis | FDA,最后访问时间:2022年2月20日。

