




随着现代互联网、信息采集以及人工智能算法等技术的创新发展和移动终端的广泛普及,数字化产品融入人们生活的各个方面,这一影响也体现在医疗服务和疾病诊疗领域。2017年,美国食品药品监督管理局(Food and Drug Administration, FDA)批准了第一个数字疗法处方类产品reSET[1]。2020年新冠疫情爆发,美国卫生与公众服务部(United States Department of Health and Human Services, HHS)和FDA联合发布了《用于在新冠疫情期间治疗心理疾病的数字健康设备的实施政策》[2],加快了数字疗法(Digital Therapeutics,DTx)紧急审批的进程[3]。根据相关统计数据,截至2022年4月,已有约40款数字疗法产品获得FDA审批。[4]据MarketsandMarkets预计[5],全球数字疗法市场规模将从2021年的34亿美元于2026年快速增长至131亿美元,复合年均增长率可达到31.4%。
一、数字疗法的概念
数字疗法(Digital Therapeutics, DTx)的概念来源于2017年成立的数字疗法联盟(Digital Therapeutics Alliance, DTA)所发布的《2018年数字疗法白皮书》[6](Digital Therapeutics Industry Report 2018)。DTA根据功能覆盖范围和临床证据要求,将数字化健康产品(DTx Product)分为数字健康(Digital Health)、数字诊疗(Digital Medicine)[7] 和数字疗法(Digital Therapeutics)三类。其中“数字健康”概念最为宽泛,包括一切能够帮助消费者改善生活方式和健康相关的产品,如大健康类手机APP、互联网医院平台等。“数字诊疗”产品则是数字化健康产品中包含检查和/或干预功能的用于医疗流程的技术或产品。“数字疗法”则是范围最小的定义,根据DTA的定义,数字疗法系由高质量软件程序所驱动,基于循证医学的疾病干预措施,用以预防、管理或治疗疾病(Digital therapeutics (DTx) deliver evidence-based therapeutic interventions that are driven by high quality software programs to prevent, manage, or treat a medical disorder or disease)[8]。数字疗法在现有传统的药物治疗、非药物治疗(手术、放射和物理治疗)及心理行为治疗方法外提供了一种新的治疗方式,其可以单独使用,也可以与药物、医疗器械或其他疗法配合使用,主要通过信息(如文字、图片、视频、音频等)、物理因子(如光线、声音、振动、磁场及其组合等)、药物等对患者施加影响,以达到优化患者护理和干预疾病的目的。
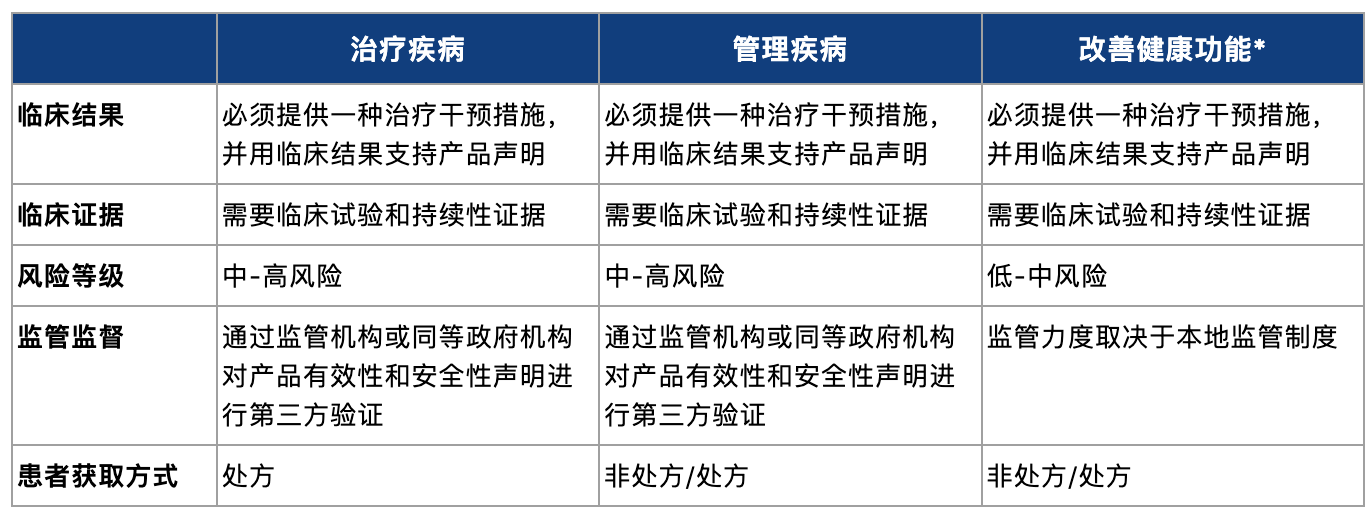
根据DTA的分类标准,符合行业核心原则的数字疗法产品按照主要用途可分为治疗、管理、预防改善三个大类,各类的特征如下[9]:
二、美国对数字疗法的监管
以美国日益发展的数字医疗监管环境为示例,数字疗法产品在美国受到FDA的监管,并由FDA下属负责管理医疗器械部门医疗设备与放射健康中心(Center for Devices and Radiological Health, CDRH)开展具体工作。特别地,FDA在CDRH下还组建了数字健康卓越中心(Digital Health Center of Excellence, DHCoE),旨在加强数字医疗的监管工作。
FDA通常将数字疗法产品归入医疗器械中的软件类产品(SaMD, Software as a Medical Device)类别进行监管,国际医疗器械监管者论坛(IMDRF, International Medical Device Regulators Forum)关于SaMD给出的定义是:SaMD具有一个或多个医疗用途,是无需医疗器械硬件即可完成预期医疗用途的软件(Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device)。在此分类逻辑下,数字疗法的监管基本与传统医疗器械的管理无异,即根据风险等级和管理程度分成3类进行上市前管理。目前多数数字疗法产品属于II类医疗器械[10],需进行上市前通告(Premarket Notification, PMN)(即510k),还有少部分数字疗法产品会被认定为III类医疗器械,需获得上市前许可(Premarket Approval, PMA)方可上市销售。
需要注意的是,数字疗法仍是一个新兴市场或产品的概念,不能将其直接与SaMD等同起来。FDA也意识到传统的监管框架并不是为数字疗法产品量身定做的,将数字疗法产品依照医疗器械法律法规进行监管的前提需满足FDA对医疗器械的定义,即由生产者设计为下列目的用于人体的,不论是单独使用还是组合使用,包括使用所需软件在内的任何仪器、设备、器具、材料或者其他物品:目的是为了疾病的诊断、预防、监护、治疗或缓解,或是意在影响身体的组成部分或功能[11]。通常数字疗法产品会比传统医疗器械需要更多实质性临床实验数据与证据证明。同时,需要何种证据才能进入市场也是一大问题,FDA认为,如人工智能/机器学习(AI/ML)驱动的产品不同于传统SaMD,会带来分类及监管上的问题。为此FDA相应地设立了“软件类产品提前审查试点项目”(Software Precertification Pilot Program, Pre-Cert)专门协助机构搭建基于软件基础发展的新兴医疗器械监管框架。此外,美国《21世纪治愈法案》(21st Century Cures Act)[12]的出台排除了将例如医疗机构综合治理技术支持软件、帮助人们保持健康习惯或用于病人电子档案记录的部分软件产品作为医疗器械进行监管,进一步对FDA的管辖范围作出明确。
三、中国对数字疗法的监管
(一)数字疗法产品是否属于医疗器械?
数字疗法仍是一个新兴的市场与产品概念,因此国内在法律、法规层面上尚未明确定义数字疗法这一概念。根据《医疗器械监督管理条例》规定,医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件,其效用主要通过物理等方式获得,目的在于疾病的诊断、预防、监护、治疗或者缓解等。从这个定义出发,符合医疗器械定义的数字产品应当归入医疗器械。《医疗器械分类目录》(2018年8月1日实施,不时动态调整)也明确将独立医用软件列为第21类别。可见,在管理属性方面,目的是预防、管理与治疗疾病的数字疗法产品在我国目前的监管语境下存在将其界定为医疗器械的可能。
国家药品监督管理局(NMPA)分别于2017年12月和2021年7月发布《关于发布移动医疗器械注册技术审查指导原则的通告》(简称“《移动医疗器械注册技术审查指导原则》”)与《关于发布人工智能医用软件产品分类界定指导原则的通告》(简称“《人工智能医用软件产品分类界定指导原则》”),提出“移动医疗器械”和“人工智能医用软件”两个与DTA对数字疗法的定义较为相似的概念。“移动医疗器械”是指采用无创“移动计算终端”实现一项或多项医疗用途的设备和/或软件。“人工智能医用软件”是指基于医疗器械数据,采用人工智能技术实现其医疗用途的独立软件。
《移动医疗器械注册技术审查指导原则》及《人工智能医用软件产品分类界定指导原则》中均划定了移动医疗器械或AI医用软件与普通医用软件的边界,对判断特定数字疗法产品是否属于医疗器械具有借鉴意义。在预期用途及目标人群方面,《移动医疗器械注册技术审查指导原则》明确将移动计算设备或软件的预期用途及目标人群作为医疗器械属性的判定标准,即移动医疗器械应为预期用于疾病管理且具有医疗目的,其目标人群为医护人员和患者。
此后,《人工智能医用软件产品分类界定指导原则》就如何判断独立软件是否属于人工智能医用软件,从而是否纳入医疗器械管理提出三个维度的判定标准:[13]
第一,预期用途,即软件的输出信息用于疾病的诊断和治疗。与《移动医疗器械注册技术审查指导原则》相同,通过预期用途的识别将人工智能医用软件与用于医院管理、新药研发、健康管理等的软件产品进行区分。
第二,产品的处理对象(软件输入)应为医疗器械数据,即医疗器械产生的用于医疗用途的客观数据,如医学影像设备产生的医学影像数据、医用电子设备产生的生理信号数据等。特殊情形下,可包含通用设备(非医疗器械)产生的用于医疗用途的客观数据。若软件产品的处理对象为非医疗器械数据(如患者主诉等信息、检验检查报告结论),不作为医疗器械管理。
第三,产品的核心功能是对医疗器械数据的处理、测量、模型计算、分析等。若软件产品基于公开的临床指南、文献、公式等,对医疗器械数据进行简单的统计、运算等,亦不作为医疗器械管理。
数字疗法是否可以注册为药品?从中国的立法而言,似乎暂无空间。《药品管理法》中对“药品”的定义为,药品指用于预防、治疗、诊断人的疾病,有目的地调节人的生理机能并规定有适应症或者功能主治、用法和用量的物质,包括中药、化学药和生物制品等。从这个定义上,《药品管理法》将非物质形态(软件)存在的数字疗法产品排除在药品的大门之外。
(二)数字疗法产品的医疗器械分类
目前国家对医疗器械按照风险程度从低到高分为三类,实行分类管理,医疗器械应取得相应的医疗器械注册证或备案证明,相关的生产经营也需取得相应的医疗器械生产经营的资质并符合医疗器械生产经营质量管理规范的要求。
根据《医疗器械分类目录》,医用独立软件属于第21类,常见的数字疗法产品主要涉及第21类项下的决策支持软件(分类编码21-04,包含计算机辅助诊断/分析软件等)和其他软件(分类编码21-06,包含康复训练软件)。
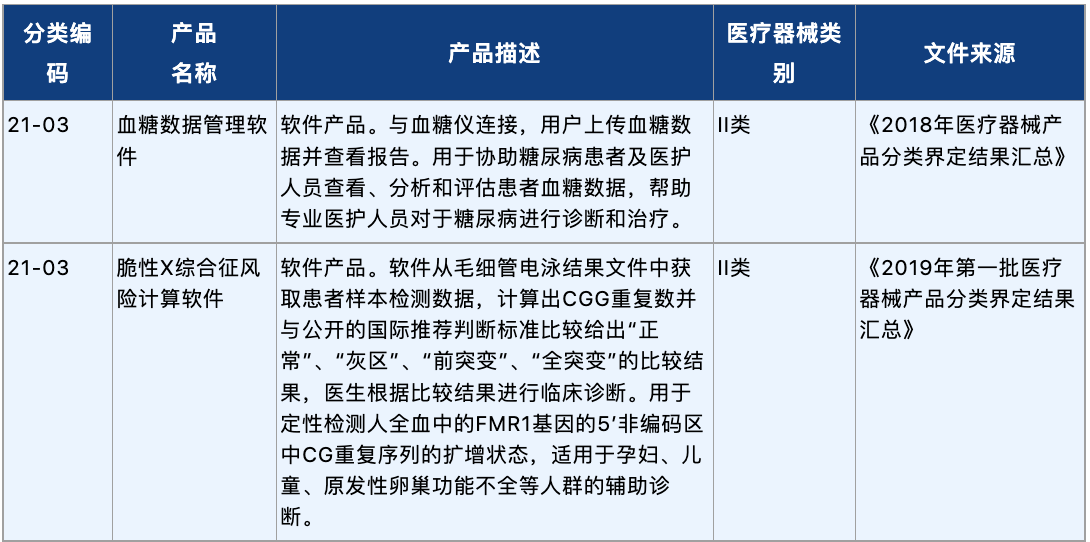
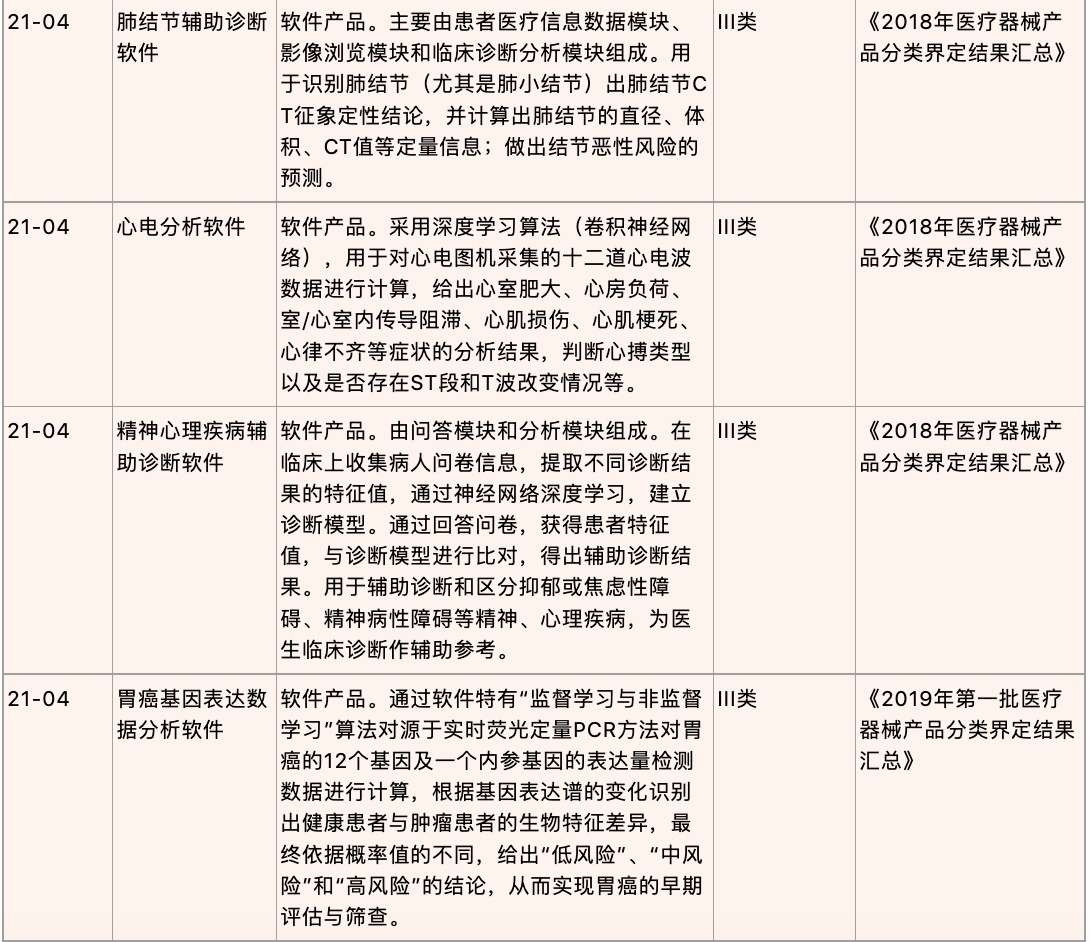
《医疗器械分类目录》明确说明,诊断功能软件风险程度按照其采用算法的风险程度、成熟程度、公开程度等为判定依据,不仅仅依据处理对象(如:癌症、恶性肿瘤等疾病的影像)为判定依据。若诊断软件通过其算法提供诊断建议,仅具有辅助诊断功能,不直接给出诊断结论,则按照第II类医疗器械管理。若诊断软件通过其算法对病变部位进行自动识别,并提供明确的诊断提示,则其风险级别相对较高,按照第III类医疗器械管理[14]。此外,根据2018年-2020年中国食品药品检定研究院(中检院)发布的分类界定汇总(汇总节选见下文),辅助诊疗功能的医疗软件产品通常建议按照决策支持类软件,即III类医疗器械进行管理。而治疗康复功能的医疗软件产品建议的产品分类则涵盖II类与III类医疗器械,需结合软件产品的具体情形进行判定。
以下是中检院关于数字疗法相关产品的分类界定节选,可以更具体地反映不同分类项下产品的特质,以便于产品进行注册申报时作为比较全面的参考:
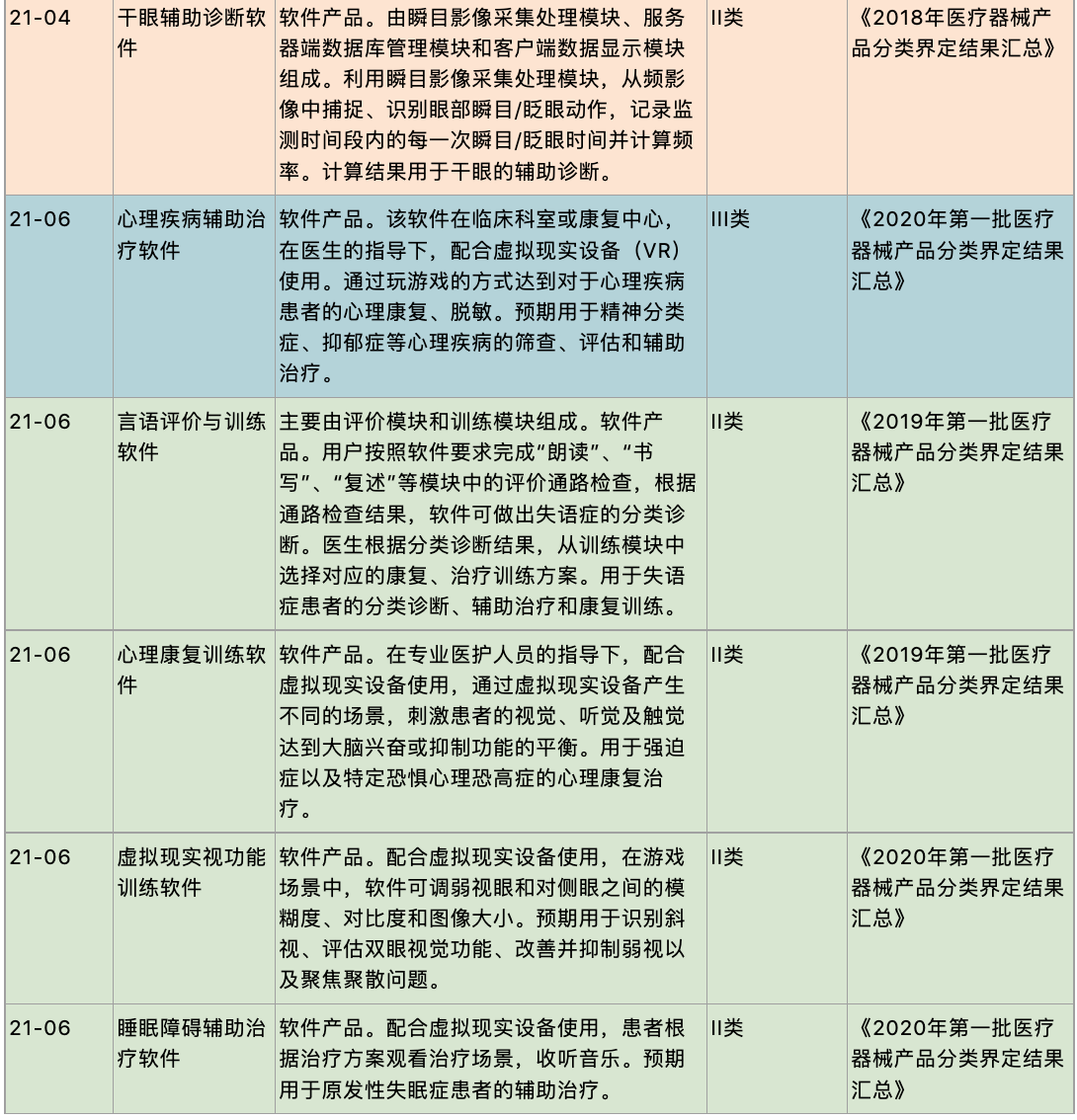
(三)临床评价
根据《医疗器械注册与备案管理办法》(国家市场监督管理总局令第47号),除可以免于临床评价的特殊情形外,医疗器械产品注册、备案通常应当进行临床评价,对临床数据进行分析、评价以确认医疗器械在适用范围内的安全性与有效性。数字疗法产品中,根据《免于临床评价医疗器械目录》(国家药监局通告2021年第71号),可以免于临床评价的特殊情形包括第21类医疗独立软件中的迹法分析软件、临床管理软件等。而具体的产品是否落入前述免于临床评价的范围内,建议综合算法应用等多种因素考虑并咨询专业机构的意见。
根据《医疗器械注册与备案管理办法》,开展医疗器械临床评价可以分为(1)开展临床试验,或(2)通过对同品种医疗器械临床文献资料、临床数据进行分析评价。当已有临床文献资料、临床数据不足以确认产品安全性、有效性时,应当开展临床试验。
通过同品种医疗器械临床文献资料、数据进行临床评价的,临床评价资料应当包括申请注册产品与同品种医疗器械的对比,同品种医疗器械临床数据的分析评价与申请注册产品与同品种产品存在差异时的科学证据等。根据《医疗器械临床试验设计指导原则》(食品药品监管总局通告2018年第13号),对照试验应优先采用疗效和安全性已得到临床公认的已上市同类产品,如因合理理由不能采用已上市同类产品,可选用尽可能相似的产品作为阳性对照。由于数字疗法仍处在小众领域,同品种产品的范围较小,受限于等同医疗器械的临床数据或可比医疗器械的临床数据的相对稀缺性,在未来的一段时间内数字疗法产品可能多需以临床试验的方式进行临床评价。
(四)注册与审批
数字疗法产品的注册审批流程与普通医疗器械大体相同,但由于医疗器械软件产品没有物理实体,且相较于传统的医疗器械更新迭代迅速,医疗器械软件在监管方面有其特殊性。
根据《医疗器械软件注册审查指导原则(2022年修订)》(简称“《指导原则》”),医疗器械软件需要考虑其更新过程中的风险管理、质量管理和软件工程的要求保证其安全性与有效性。注册时,制造商应基于软件安全性级别提交相应注册申报的资料。此外,《指导原则》基于对医疗器械软件的安全性和有效性的影响,将软件更新分为重大软件更新及轻微软件更新两大类。相应地,由于软件更新管理只能通过状态标识进行,软件版本又可分为软件发布版本和软件完整版本,软件发布版本仅体现重大软件更新,其改变意味着发生重大软件更新,医疗器械软件应申请变更注册;软件完整版本发生改变但软件发布版本未变表示软件仅发生轻微更新,此时通过质量管理体系进行控制,无需申请变更注册。具体到命名规则方面,例如,软件版本命名规则为X.Y.Z.B,其中X表示重大增强类软件更新,Y表示轻微增强类软件更新,Z表示纠正类软件更新,B表示软件构建,则软件发布版本为X,软件完整版本为X.Y.Z.B,此时X改变应申请变更注册,而Y、Z、B改变但X未变则无需申请变更注册。
(五)数据合规
近年来,《网络安全法》《数据安全法》和《个人信息保护法》共同构建了我国的网络数据安全治理体系。数字疗法产品通常包括医疗服务过程中产生的大量个人信息和疾病防治、健康管理相关的信息等数据,随着我国网络安全和个人信息监管体系的日渐完善,数字疗法相关的数据合规的重要性随之提升。
一方面,数字疗法产品相关企业在业务开展与运营过程中,针对直接面向患者人群的数字疗法产品,应当公开个人信息的处理规则,明确明示处理信息的特定目的、目的的充分必要性,如疾病的诊断、预防或治疗等,同时需要健全数据安全的管理制度,采取相应的技术措施或必要措施保障数据的安全。发现数据安全缺陷或漏洞等风险时,应立即采取相应的补救与处置措施。涉及重要数据的,重要数据的处理者应当按照规定对其数据处理活动定期开展风险评估,并向有关主管部门报送风险评估报告。
2017年,NMPA特别制定颁布了《医疗器械网络安全注册技术审查指导原则》(简称“《医疗器械网络安全指导原则》”)。《医疗器械网络安全指导原则》要求具有网络连接功能以进行电子数据交换或远程控制以及采用存储媒介以进行电子数据交换的II类、III类医疗器械产品的注册人应当在医疗器械全生命周期过程(包括设计开发、生产、分销、部署、维护)中保证医疗器械产品自身的网络安全,从而保证其安全性和有效性。医疗器械软件若具备电子数据交换、远程访问与控制、用户访问三种功能当中一种及以上功能,均需考虑网络安全问题。
另一方面,健康医疗数据在宏观层面上包括传统医疗领域中从事医疗活动产生的检查与治疗等相关数据,也包括在线平台、大数据应用、数字疗法这样类似的新兴技术产品与传统医疗活动结合产生的新型数据如电子病历、智能监测生命体征甚至遗传基因数据等。传统上,医疗机构和数字疗法后台运营方作为健康医疗大数据的主要采集和存储方,根据国家卫生健康委员会(前身为国家卫生和计划生育委员会)于2014年5月5日颁布的《人口健康信息管理办法(试行)》(国卫规划发〔2014〕24号),其采集、利用、管理人口健康信息应当遵循医学伦理原则,保证信息安全,保护个人隐私,按照“一数一源、最少够用”的原则采集人口健康信息,将人口健康信息实行分级存储与管理,按照个人和单位授权要求进行利用,且责任单位应当建立痕迹管理制度,任何建立、修改和访问人口健康信息的用户都应当通过严格的实名身份鉴别和授权控制。
根据《关键信息基础设施安全保护条例》对“关键信息基础设施”作出的定义[15],参照《国家网络安全检查操作指南》认定步骤,收集并存储超过100万个人信息的医疗机构和数字疗法后台运营方可能被归入“关键信息基础设施运营者”的范围。根据《关键信息基础设施安全保护条例》,“关键信息基础设施运营者”承担一系列网络安全合规义务,其应当依照法律法规及国家标准强制性要求在网络安全等级保护的基础上采取技术保护措施和其他必要措施,设置专门安全管理机构,自行或者委托网络安全服务机构对关键信息基础设施每年至少进行一次网络安全监测和风险评估,并向监管部门、公安机关报告关键信息基础设施发生的重大网络安全事件或发现的重大网络安全威胁等。实践中,监管部门可结合行业实际与认定规则,负责组织认定本行业领域关键信息基础设施,并将认定结果通知运营者。医疗机构和数字疗法后台运营方在提供数字医疗产品服务的过程中,应当密切关注法律法规与监管通知,做好合规方面的预判。
四、展望
数字疗法将循证医学和软件科技结合,拓展了心理、神经类疾病治疗手段,其商业路径逐步得到验证。从监管角度展望,美国在经过早期探索后,针对数字疗法的特点设立了单独的审批目录和流程。在中国,对数字疗法产品目前依照医疗器械软件(包括作为医疗器械或其附件的独立软件和作为医疗器械或其部件、附件组成的软件组件)的相关政策和流程进行审批和监管。数字疗法仍缺乏针对性的官方定义、行业标准、临床试验和监管政策。数字疗法的兴起为监管提出新的挑战和课题。我们期待NMPA逐步填补规则空隙,监管规则的逐步完善将为方兴未艾的数字疗法铺就更坚实的基础。
注释:
[1] https://peartherapeutics.com/products/reset-reset-o/,最后访问日期2022年8月1日。
[2] Enforcement Policy for Digital Health Devices For Treating Psychiatric Disorders During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/enforcement-policy-digital-health-devices-treating-psychiatric-disorders-during-coronavirus-disease,最后访问日期2022年8月1日。
[3] 如“用于心理健康疾病的数字化行为疗法设备及其他设备”和“针对心理健康和精神状态治疗的低风险全面健康护理和数字健康产品”,只要求软件检测和网络安全在内的质量评估,并明确标注产品医用说明、适应症、适用年龄、严重程度和临床试验简述等即可紧急审批该等产品为人们提供服务。
[4] Nidheesh Kesav, Srinivas H , Eric Walker & Santanu Sen, Prescription digital therapeutics (2022), https://www.virtusa.com/perspectives/article/prescription-digital-therapeutics. 最后访问日期2022年8月1日。
[5] MarketsandMarkets,《全球数字疗法市场规模》。
[6] https://dtxalliance.org/2018/10/23/dtaindustryreport2018/,最后访问日期2022年8月1日。
[7] 国内的翻译表述多有不一致,Medicine即可是狭义的药物,也可以是广义的医学概念。此处译为诊疗考虑到要区分于Digital Health,且仅译为药物(本是干预疗法的一种)范围过于狭窄。
[8] https://dtxalliance.org/wp-content/uploads/2021/01/DTA_DTx-Definition-and-Core-Principles.pdf,最后访问日期2022年8月1日。
[9] https://dtxalliance.org/wp-content/uploads/2021/01/DTA_FS_DTx-Product-Categories_010521.pdf,最后访问日期2022年8月1日。
[10] 例如Pear Therapeutics公司针对慢性失眠症开发的产品Somryst、Akili Interactive公司针对注意力缺陷多动障碍(ADHD)儿童开发的视频游戏产品Endeavor Rx均属于II类医疗器械。
[11] 21 U.S. Code § 321(h).
[12] Section 3060(a) of the 21st Century Cures Act (Cures Act).
[13] 余新华、戎善奎、彭亮:《AI医疗软件属于医疗器械吗?专家提醒:必须符合这三大条件》,中国医药报。
[14] 《医疗器械分类目录》:第21节 医用软件说明。
[15] 《关键信息基础设施安全保护条例》第二条:本条例所称关键信息基础设施,是指公共通信和信息服务、能源、交通、水利、金融、公共服务、电子政务、国防科技工业等重要行业和领域的,以及其他一旦遭到破坏、丧失功能或者数据泄露,可能严重危害国家安全、国计民生、公共利益的重要网络设施、信息系统等。
